Please enable JavaScript in your browser to see this interactive content.
Referral and follow up
All patients should have access to a local cardiologist and should also receive at least one initial assessment at a specialist CHD centre. Adults with moderate or severely complicated CHD should have their care shared between their local centre and a specialist CHD centre.
Patients with CHD require lifelong follow up. Emphasis is on quality and longevity of life rather than cure.
Treatment options
- Surgical Management: Many patients receive surgery when they are babies or during childhood. For some patients this involves multiple surgeries. There are very few operations that offer a “cure” for their heart defect and instead these are described as palliative operations or repairs. As adults, the decision to offer surgery depends on a number of factors. Patients require regular reviews to look for changes in their symptoms, haemodynamic status or exercise tolerance, and reviewing echo and MRI imaging for evidence of changes in their heart. Some of these operations include valve replacements or repairs, aortic root replacements, coractation repairs, closing ASD’s or VSD’s and. in some cases, heart or heart and lung transplantation.
- Trans Catheter: There have been many advances in transcatheter interventions in recent years which provide an opportunity for some patients to receive a much less invasive treatment. For example, some ASDs, VSDs, and PDAs can be closed with devices and some coarctations of the aorta and stenosed pulmonary arteries can be stented, all through a transcatheter approach.
Complications
Heart failure
Heart failure is a common problem for CHD patients and, due to the lack of research into heart failure specifically in the congenital context, ESC guidelines suggest that patients are treated in line with current, general heart failure management recommendations. However, the effectiveness of these treatments (particularly for patients with systemic right ventricles or Fontan circulations) is unknown.
Arrhythmias
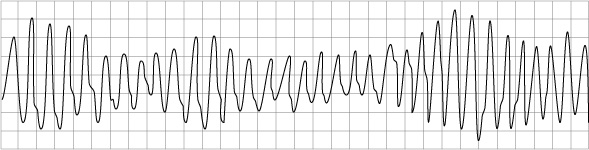
Arrhythmias are the main reason for hospitalisation in ACHD patients. Arrhythmias are often a sign of haemodynamic deterioration in CHD patients and they often need urgent Direct Current Cardioversion (DCCV), particularly in the case of patients who have Fontan circulations. Antiarrhythmic drug therapy is often poorly tolerated due to the negative inotropic effects.
Individuals with unexplained syncope (particularly in patients with Tetralogy of Fallot; Transpostion of the Great Arteries and congenitally corrected Transposition of the Great Arteries; Univentricular hearts; and Aortic Stenosis) are at highest risk of sudden cardiac death.
Pulmonary Arterial Hypertention (PAH)
The development of PAH occurs in patients who have untreated systemic to pulmonary shunts (blood flowing through a defect in the heart from the left side to the right side of the heart).
In cases such as Eisenmenger’s Syndrome this shunting process eventually leads to PAH and causes irreversible damage to the lungs which then, in turn, results in a reversal of the shunting. This reversal (blood flowing from the right side of the heart, through a defect and into the left side of the heart) results in the patient becoming cyanosed.
Patients who have PAH in the context of CHD can be offered drug therapies, which need to be managed by a specialist congenital centre. These medications include prostanoids, endothelin receptor antagonists (ERAs) and phosphodiesterase type-5 inhibitors.
Patients with PAH and cyanosis require complex management and the condition will involve multiple organs. Some factors to consider (as per ESC guidelines):
- High risk of both clotting and bleeding. High risk of cerebral events and abscesses. Should receive prophylactic antibiotics for high risk dental procedures.
- Important to keep hydrated and avoid acute exposure to heat such as saunas, hot tubs etc. Avoid iron deficiency and anaemia, respiratory infections and agents that can lead to renal impairment.
- Avoid pregnancy, moderate to severe strenuous exercise and high altitude.
- Use air filters in IV lines to prevent air embolisms, as high risk of entering system.